A case of SUDEP in a patient with Dravet syndrome with SCN1A mutation
*Franc¸ois Le Gal, yChristian M. Korff, *Christine Monso-Hinard, zMichael T. Mund,
*Michael Morris, *xAlain Malafosse, and {Thomas Schmitt-Mechelke
*Genetic Medicine, University Hospitals of Geneva, Geneva, Switzerland; yPediatric Neurology, Pediatric Specialties Service, Child and Adolescent Department, University Hospitals of Geneva, Geneva, Switzerland; zInstitute of Legal Medicine, University of Zurich, Zurich, Switzerland; xDepartment of Psychiatry, University of Geneva, Geneva, Switzerland; and {Department of Childhood Neurology, Children’s Hospital, Lucerne, Switzerland
SUMMARY
A boy with a clinical history of pharmacologically resistant Dravet syndrome died suddenly after falling asleep. The autopsy concluded that the cause of death was sudden unexpected death in epilepsy (SUDEP). Postmortem molecular analysis of the SCN1A gene by multiplex ligation-dependent probe amplification (MLPA), high-resolution melting curve analysis (HRMCA), and sequencing revealed a frameshift duplication of adenosine at position 504. The incidence of this mutation is discussed as a potential cause of SUDEP.
KEY WORDS:
SUDEP, Dravet syndrome, SMEI, SCN1A. Sudden unexpected death in epilepsy (SUDEP) is a relatively rare event, but a major problem for people with epilepsy. The likelihood of this dramatic event ranges from 0.09 to 0.35 per 1,000 person-years in population-based studies to as many as _1 per 100 person-years in surgical cohorts of patients with intractable epilepsy. SUDEP is likely to be the consequence of an interaction between vulnerability factors, such as genetic factors, and a cascade of developmental, clinical, and environmental factors. Many clinical risk factors for SUDEP have been identified, the most important of which are pharmacoresistance with frequent seizures, history and number of generalized tonic– clonic seizures (GTCS), early epilepsy onset, high number of antiepileptic drug (AED) changes, and a full-scale IQ of <70 (reviewed by So, 2008). The clearest risk factor is a history of generalized tonic–clonic seizures (GTCS) and uncontrolled epilepsy (Nashef & Ryvlin, 2009).
A genetic susceptibility to SUDEP has been recently suggested, but very few studies have been published on this
hypothesis. To date, only one mutation (R523C) in the cardiac voltage-gated Va subunit sodium channel (Nav1.5, or SCN5A) has been reported in a patient who died from SUDEP (Aurlien et al., 2009). Hindocha et al. (2008) recently described a family with generalized epilepsy and febrile seizures plus (GEFS+), in which two affected patients died from SUDEP. A mutation (I1867T) in the gene coding the sodium channel subunit 1 (SCN1A, MIM 182389) was identified in all affected relatives, but no DNA samples from SUDEP cases were available. We report here the first SCN1A mutation in a patient with Dravet syndrome (severe myoclonic epilepsy of infancy) and cortical dysplasia, who died from definite SUDEP.
Methods
Informed written consent was obtained from the patient’s mother before genetic analysis, as required by the Swiss law.
Patient history and autopsy:
The male patient was the first offspring of a healthy mother with a negative family history for epilepsy; no medical information was available about the father. The patient presented with a prolonged febrile seizure at the age of 5 months, which was followed by recurrent febrile and afebrile generalized tonic–clonic as well as partial seizures with secondary generalization and atonic seizures despite anticonvulsant treatment with vitamin B6, valproic acid, carbamazepine, and vigabatrin. Cerebral magnetic resonance imaging (MRI) (at the age of 21 months), workup for neurometabolic diseases, and karyotype were all normal. Several electroencephalography (EEG) recordings in infancy showed generalized slowing and increase of theta rhythms. Electrocardiography (ECG) recordings showed normal supraventricular rhythms with normal PQ and QT intervals. At the age of 5 years, isolated myoclonic seizures and generalized spike-wave discharges were documented on EEG. The clinical diagnosis of Dravet syndrome was then made. In the following years, generalized and complex partial seizures persisted, often occurring in clusters, despite treatment with topiramate, levetiracetam, and clobazam. Cognitive development was normal in the first 2 years of life but showed a dementia-like decline in the following years resulting in severe mental retardation (at 5.5 years of age his developmental quotient was evaluated to be 40) with specific impairment of language development. At the age of 11 years, while being treated with lamotrigine (10 mg/kg) and levetiracetam (50 mg/kg), the boy was found dead by his mother in a supine position after having fallen asleep. Attempts at resuscitation were unsuccessful. Forensic examination including autopsy, radiographic, microbiologic, virologic, and chemical-toxicologic studies revealed no obvious cause of death. Neuropathologic examination detected multifocal micronodular dysplasia of the left temporal cortex, bilateral gliosis of the hippocampal CA4 region, and global brain edema without mass effect. No typical signs of prolonged status epilepticus were seen.
Genomic DNA preparation and mutation analyses
Genomic DNA was extracted from frozen blood, harvested, and purified with Qiagen columns (QIAamp DNA Blood Mini Kit; Qiagen GMBH, Hilden, Germany). Multiplex ligation-dependent probe amplification (MLPA) was performed using Salsa MLPA P137 SCN1A reagent (MHRC-Holland, Amsterdam, The Netherlands). Quantification analysis was carried out using GeneMarker software v1.6 after separation of polymerase chain reaction (PCR) products by capillary electrophoresis on an ABI 3100 DNA Analyzer (Applied Biosystems, Foster City, CA,U.S.A.).
Prescreening for potential DNA mutation was carried out by high-resolution melting curve analysis (HRMCA) performed on a LightCycler 480 (Roche Applied Science, Mannheim, Germany). The LC480 High Resolution Melting Master Kit (Roche) was used according to the manufacturer’s recommendations. Patient DNA was tested in parallel with DNA from control patients. Normalized and temperature-shifted difference plots were compared between samples.
Amplicons showing abnormal HRMCA profiles were sequenced with the Big Dye Terminator Cycle Sequencing Kit v3.1 (Applied Biosystems), according to the manufacturer’s recommendations, and analyzed on capillary electrophoresis with the ABI 3100 DNA Analyzer using primers designed to cover all exons and their flanking sequences. Sequences were analyzed with GeneSearch v3.6 software. Amplicon for exon 4 was further cloned with TOPO TA Cloning kit (Invitrogen, Carlsbad, CA, U.S.A.) in TOP10 competent cells according to the manufacturer’s recommendations and eight clones were amplified, plasmid isolated with GenElute HP Plasmid Miniprep kit (Sigma, St Louis, MI, U.S.A.), and later sequenced as previously described.
Results
MLPA analysis performed in triplicate did not reveal the existence of any exon deletion (data not shown).
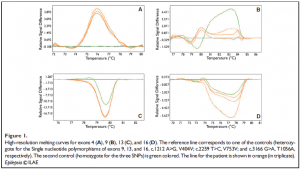
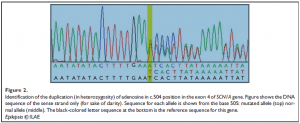
HRMCA performed three to five times per amplicon revealed constant and significant differences for exon 4 (patient vs. both controls), and for exons 9, 13, and 16 (patient vs. one of the two controls) (Fig. 1). Sequencing revealed that the differences observed in exons 9, 13, and 16 were due to the frequent polymorphisms already described (NCBI, SCN1A SNP Genereview report), as rs7580482 (G>A, V404V), rs6432860 (C>T, V753V), and rs2298771 (A>G, T1056A), respectively (data not shown). The frequencies of these polymorphisms in the general population are 0.42, 0.32, and 0.33, respectively. For exon 4, sequencing revealed a duplication of the adenosine residue in position 504, in heterozygosity (Fig. 2). Heterozygosity was confirmed by sequencing eight clones isolated from PCR product. The patient’s genotype is thus c. [504dupA] + [=].
Discussion
The mutation we found (c. [504dupA]) has not been previously reported in the SCN1A variant database (Claes et al., 2009). As a frameshift, this duplication is likely to impact severely the function of the SCN1A protein. In the clinical context, this result is consistent with Dravet syndrome. Several SCN1A exonic deletions and SCN1A mutations have already been reported in this syndrome (Mulley et al., 2006; Depienne et al., 2009), as well as in generalized epilepsy with febrile seizures plus (GEFS+, Escayg et al., 2000) and in familial hemiplegic migraine (Dichgans et al., 2005).
SCN1A mutations are present in at least 80% of patients with Dravet syndrome, and the incidence of SUDEP is higher in Dravet syndrome (Dravet et al., 2005) than in most forms of severe epilepsy. The association we report here, therefore, could be considered to be a coincidence. However, SCN1A mutations and SUDEP cases have been previously reported in various families, suggesting that there may be a direct link between the two. In the case of a GEFS+ family, it has been suggested that an SCN1A mutation was associated with SUDEP, but no genetic testing of the two patients who died from SUDEP was performed (Hindocha et al., 2008).
To the best of our knowledge, this is the first case of an SCN1A mutation in a Dravet syndrome patient who died from SUDEP. The role of SCN1A mutations in SUDEP remains unclear. According to animal research findings, it would appear that some mutations lead to abnormal expression of SCN1A in the heart and consequently to cardiac arrhythmias. Although SCN1A is primarily expressed in the brain, its product was also shown to be present in the heart in rats, mice, and dogs (cited by Hindocha et al., 2008). SCN5A mutations are associated with Brugada syndrome (Tfelt-Hansen et al., 2010), and with sudden infant death among African Americans (Plant et al., 2006). Future studies should explore SCN5A and other long-QT syndromeassociated genes to test the hypothesis that several mutations are combined in this fatal event. Such cases must be systematically reported to allow future evaluations of the relative risk of SUDEP in Dravet syndrome patients with specific types of SCN1A mutations and also in SUDEP patients other than Dravet syndrome.
Acknowledgments
This study was partly financed by grants (UCB Pharma AG, Swiss Life Foundation, Pictet & Cie, and Prim’Enfance Foundation) to CMK. We are most grateful to Dr. Philippe Maillet (Molecular Oncology, University Hospitals of Geneva) for his help in setting up the SCN1A HRMCA test.
Disclosure
None of the authors has any conflict of interest to disclose. We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
References
Aurlien D, Leren TP, Taubøll E, Gjerstad L. (2009) New SCN5A mutation in a SUDEP victim with idiopathic epilepsy. Seizure 18(2):158–160.
Claes L, Deprez L, Suls A, Baets J, Smets K, Van Dyck T, Deconinck T, Jordanova A, De Jonghe P. (2009) The SCN1A variant database: a novel research and diagnostic tool human mutation database. http://www.molgen.ua.ac.be/SCN1AMutations.
Depienne C, Trouillard O, Saint-Martin C, Gourfinkel-An I, Bouteiller D, Carpentier W, Keren B, Abert B, Gautier A, Baulac S, Arzimanoglou A, Cazeneuve C, Nabbout R, LeGuern E. (2009) Spectrum of SCN1A gene mutations associated with Dravet syndrome: analysis of 333 patients. J Med Genet 46(3):183–191.
Dichgans M, Freilinger T, Eckstein G, Babini E, Lorenz-Depiereux B, Biskup S, Ferrari MD, Herzog J, van den Maagdenberg AM, Pusch M, Strom TM. (2005) Mutation in the neuronal voltage-gated sodium channel SCN1A in familial hemiplegic migraine. Lancet 366(9483):371– 377.
Dravet C, Bureau M, Oguni H, Fukuyama Y, Cokar O. (2005) Severe myoclonic epilepsy in infancy (Dravet syndrome). In Roger J, Bureau M, Dravet C, Genton P, Tassinari C, Wolf P (Eds) Epileptic syndromes in infancy, childhood and adolescence, 4th ed. John Libbey, London, pp. 77–89.
Escayg A, MacDonald BT, Meisler MH, Baulac S, Huberfeld G, An-Gourfinkel I, Brice A, LeGuern E, Moulard B, Chaigne D, Buresi C, Malafosse A. (2000) Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nat Genet 24(4):343–345.
Hindocha N, Nashef L, Elmslie F, Birch R, Zuberi S, Al-Chalabi A, Crotti L, Schwartz PJ, Makoff A. (2008) Two cases of sudden unexpected death in epilepsy in a GEFS+ family with an SCN1A mutation. Epilepsia 49(2):360–365.
Mulley JC, Guerrero NS, Dibbens L, Iona X, McMahon JM, Harkin L, Schouten J, Yu S, Berkovic SF, Scheffer IE. (2006) A new molecular mechanism for severe myoclonic epilepsy of infancy: exonic deletions in SCN1A. Neurology 67:1094–1095.
Nashef L, Ryvlin P. (2009) Sudden Unexpected Death in Epilepsy (SUDEP): update and reflections. Neurol Clin 27:1063–1074.
Plant LD, Bowers PN, Liu Q, Morgan T, Zhang T, State MW, Chen W, Kittles RA, Goldstein SAN. (2006) A common cardiac sodium channel variant associated with sudden infant death in African Americans, SCN5A S1103Y. J Clin Invest 116(2):430–435.
So EL. (2008) What is known about the mechanisms underlying SUDEP? Epilepsia 49(suppl 9):93–98. Tfelt-Hansen J, Winkel BG, Grunnet M, Jespersen T. (2010) Inherited cardiac diseases caused by mutations in the Nav1.5 sodium channel. J Cardiovasc Electrophysiol 21:107–115.